L’expérience clinique montre que l’utilisation des anticorps monoclonaux n’est pas dénuée d’effets toxiques chez les patients traités. Les exemples anciens comme le muromonab ou OKT- 3 (anti-CD3), ou plus récents comme le TGN1412 (anti-CD28) montrent que la prédiction des effets potentiellement délétères de ces thérapeutiques ciblées reste complexe. L’ensemble des effets toxiques observés en clinique peut être classifié en quatre types : le syndrome de libération de cytokines, l’induction de pathologies auto-immunes, la toxicité d’organe et les infections opportunistes.
L’immunogénicité des anticorps, qui est variable en fonction de leur degré d’humanisation, contribue aussi à des effets délétères sous la forme par exemple de complexes immuns. L’accident récent observé avec le TGN1412 montre bien que la relative confiance en termes de sécurité d’emploi liée à l’utilisation des anticorps en thérapeutique n’est plus d’actualité et révèle aussi les limites des modèles non cliniques en termes de prévision. Une meilleure connaissance des cibles de ces anticorps et de leurs mécanismes d’action et l’identification des facteurs de risque (activation des cellules du système immunitaire, cibles à effets pléïotropes…) doivent permettre d’améliorer la sécurité d’emploi de ces produits en clinique.Les anticorps monoclonaux (Acm), par définition, permettent une reconnaissance précise et presque exclusive d’une cible. Cet avantage, d’abord exploité dans le domaine du diagnostic, a été étendu à des applications dans le domaine thérapeutique. Au-delà de l’intérêt de la reconnaissance spécifique d’une cible thérapeutique, un des autres intérêts potentiels était une limitation des effets indésirables voire toxiques par opposition aux médicaments « chimiques ». Leur spécificité, alliée au fait que ces produits sont souvent employés dans des pathologies lourdes échappant aux traitements conventionnels, leur confère très souvent un rapport bénéfices/risques élevé. Néanmoins, l’expérience montre que l’utilisation des anticorps monoclonaux en thérapeutique n’est pas dénuée d’effets toxiques chez les patients traités. Les exemples anciens comme le muromonab ou OKT-3 (anti-CD3), ou plus récents comme le TGN1412 (anti-CD28), montrent que la prédiction des effets potentiellement délétères de ces thérapeutiques ciblées reste complexe.
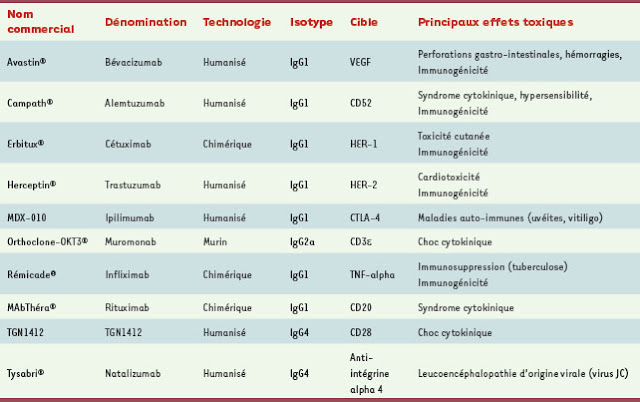
Depuis l’introduction des anticorps monoclonaux dans l’arsenal thérapeutique, l’ensemble des effets toxiques observés en clinique peuvent être regroupés en quatre types : le syndrome de libération de cytokines, l’induction de pathologies auto-immunes, la toxicité d’organes et les infections opportunistes (Tableau I). Il est aussi possible de classer ces effets toxiques en fonction du mécanisme d’action des anticorps utilisés. Dans ce cas, on distingue les effets liés à la cible, comprenant la toxicité en relation avec l’effet pharmacologique ou avec l’expression dans les tissus sains, et les effets non liés à la cible. L’immunogénicité des anticorps, qui est variable en fonction de leur degré d’humanisation, contribue aussi à des effets délétères comme la formation de complexes immuns ou l’altération des caractéristiques pharmacocinétiques (→).
Effets toxiques des anticorps monoclonaux en thérapeutique. CTLA-4: cytotoxic T-lymphocyte antigen 4 ; EGF : epidermal growth factor ; HER : human epidermal growth factor receptor ; TNF-alpha : tumor necrosis factor alpha ; VEGF : vascular endothelial growth factor.
(→) voir P. Stas et I. Lasters, page 1070
Toxicité liée à la cible
Toxicité en relation avec l’effet pharmacologique
Certaines des toxicités observées sont reliées directement aux effets pharmacologiques des anticorps sur la cible, c’est le cas des immunosuppresseurs, des inducteurs de tolérance immunitaire ou des inhibiteurs de l’angiogenèse. L’utilisation des anticorps immunosuppresseurs (natalizumab anti-intégrine α4, infliximab anti-TNFα) est associée à une augmentation des infections opportunistes dont le mécanisme est traité dans d’autres articles de ce numéro (→).
(→) voir E. Rigal et al., page 1135 ; L. Semerano et M.C. Boissier et al., page 1108 ; J. Sibilia, page 1033
Le bévacizumab (Avastin®) est un anticorps anti-VEGF (vascular endothelial growth factor) inhibiteur de l’angiogenèse [1] (→). L’angiogenèse est le processus de formation de néovaisseaux à partir de vaisseaux sanguins préexistants, il s’agit donc d’un mécanisme crucial pour le développement embryonnaire, la cicatrisation, ainsi que pour la croissance des tumeurs et des métastases. Une augmentation des perforations gastro-intestinales et des risques hémorragiques a été observée chez un nombre limité de patients traités par le bévacizumab [2]. Ces effets sont une conséquence de l’inhibition de l’activité du VEGF. Ces anomalies de cicatrisation ont aussi été observées dans les modèles animaux non cliniques [1]. Le CTLA4 (cytotoxic T-lymphocyte antigen 4) est une protéine membranaire de la famille CD28 exprimée de manière constitutive sur les lymphocytes T régulateurs [3]. L’inhibition de la fonction de CTLA4 élimine les cellules T régulatrices et augmente l’expansion et l’activation des cellules T effectrices justifiant l’emploi d’anticorps anti-CTLA4 dans les protocoles de vaccination antitumorale [3]. L’inhibition de CTLA4 par des anticorps comme l’ipilimumab (MDX-010, IgG1 humanisé) induit une augmentation de l’incidence de maladies auto-immunes comme le vitiligo ou l’uvéite. Dans une étude conduite chez 198 patients porteurs de mélanome, 21 % ont présenté une entérocolite auto-immune avec perforation intestinale nécessitant chez 5 patients une colectomie [4].
(→) voir N. Cézé et al., page 1099
Toxicité liée à l’expression de la cible dans les tissus normaux
La toxicité des anticorps thérapeutiques peut aussi s’exprimer en raison de leur interaction avec la cible exprimée sur des tissus non concernés par l’effet thérapeutique recherché (Tableau I). La toxicité cutanée du cétuximab (anti-HER-1/EGFR, IgG1, Erbitux®) et la cardiotoxicité du trastuzumab (anti-HER-2, Herceptin®) sont liées à l’expression de la cible respectivement dans la peau et dans le muscle cardiaque [5].
Le cétuximab se fixe spécifiquement sur le domaine extracellulaire du récepteur de l’EGF (epidermal growth factor) inhibant ainsi la fixation des ligands de ce récepteur (EGF, TGF-α, transforming growth factor alpha). La principale toxicité du cétuximab et des anti-HER-1/EGFR (panitumumab…) est une atteinte dermatologique de type rash acnéiforme située majoritairement sur la face, le thorax et le dos. Cette dermatotoxicité est retrouvée chez 90 % des patients traités par cétuximab avec une toxicité de grade 3 à 4 chez 16 % de ces patients [6]. Il existe une corrélation entre l’efficacité et l’incidence des rashs cutanés, qui sont liés à l’expression de HER-1 sur les kératinocytes et sur les follicules pileux.
Le trastuzumab se fixe sur le récepteur HER-2 appartenant à la famille des récepteurs EGF. HER-2 est surexprimé dans certains cancers mammaires métastatiques. En 2001, une toxicité cardiaque est décrite pour la première fois chez les patients traités par trastuzumab avec des incidences de 6 à 9 % en monothérapie, 9 à 11 % en association avec le paclitaxel1et 26 à 28 % en combinaison avec les anthracyclines2 [7]. Cette toxicité était parfaitement inattendue car HER-2 n’était pas décrit comme jouant un rôle dans la physiologie cardiaque et les études de toxicologie animale ne montraient aucune toxicité de ce type. Bien que le mécanisme de la toxicité ne soit pas complètement élucidé, les résultats obtenus chez les souris possédant une délétion du gène HER-2 au niveau cardiaque montre un effet du trastuzumab sur le rôle de HER-2 dans la survie des cardiomyocytes [8]. Une étude récente montrant une augmentation de l’expression de HER-2 chez des patients traités par les anthracyclines suggère que le système HER-2/neuréguline serait activé afin de limiter les dommages liés au stress oxydant ; en cas de traitement associant trastuzumab et anthracyclines, cet effet protecteur serait inhibé, donnant lieu à une exacerbation de la toxicité des anthracyclines [9].
Syndrome de libération de cytokines et choc « cytokinique »
Un certain nombre d’anticorps thérapeutiques provoquent une libération de cytokines pro-inflammatoires comme le TNF-α(tumor necrosis factor alpha) et l’IFN γ (interféron-γ) entraînant généralement une dyspnée, une fièvre, des frissons et parfois une urticaire. Cette symptomatologie, généralement réversible, peut être induite par le rituximab (anti-CD20, Mabthéra®) ou encore l’alemtuzumab (anti-CD52, MabCampath®) [10, 11]. Dans certains cas, la libération de cytokines est massive et soudaine, aboutissant à une défaillance des organes vitaux ressemblant à ce qui est observé dans le choc septique, il s’agit d’un choc cytokinique. Ce choc cytokinique a été décrit pour la première fois après l’administration du muromonab (anti-CD3, Orthoclone OKT3®) (→) et plus récemment avec le TGN1412 (anti-CD28) [12, 13]. Bien que la symptomatologie clinique liée à la libération de cytokines soit souvent proche de celle décrite dans les réactions d’hypersensibilité, il s’agit de mécanismes différents qui ne sont pas liés à l’immunogénicité des anticorps.
(→) voir B. Vanhove, page 1121
Les mécanismes de libération de cytokines peuvent impliquer une fixation directe de l’anticorps sur la cellule cible exprimant l’antigène (cas du muromonab et du TG1412) ou le recrutement de cellules effectrices dans le cas des anticorps d’isotype IgG1 (cas de l’alemtuzumab et du rituximab). Dans le cas du muromonab, ce serait l’activation des lymphocytes T après fixation de l’anticorps sur la molécule CD3 de manière dépendante de la région Fc qui serait responsable de la production des cytokines [12]. Dans ce cas, le fragment Fc se fixe sur un récepteur (RFcγ) situé sur une cellule autre que le lymphocyte T. Ceux des anticorps monoclonaux anti-CD3 qui ne se fixent pas sur les récepteurs au fragment Fc ne provoquent pas de choc cytokinique bien qu’une activation modérée des lymphocytes T soit toujours présente, associée à des effets secondaires modérés comme de la fièvre, des maux de tête et des troubles gastro-intestinaux [14]. Pour l’alemtuzumab, et probablement le rituximab, ce sont les cellules NK (natural killer) recrutées après fixation de la région Fc de l’anticorps sur les RFcγIII (CD16) présentes sur les cellules NK mais pas sur les cellules cibles qui seraient responsables de la libération de TNF-α [11].
Le TGN1412 appartient à une nouvelle classe d’anti-CD28 appelée CD28 « superagonistes ». Ces anticorps, dont le mécanisme d’action n’est pas encore élucidé, sont capables de provoquer l’activation des lymphocytes T de souris, de rat et humains en l’absence de stimulation du récepteur de l’antigène. Le 13 mars 2006, l’administration du TGN1412 par voie intraveineuse chez 6 volontaires sains, au cours du premier essai de phase 1 initié par la société TeGenero, a provoqué une toxicité extrêmement sévère chez tous les volontaires recevant le produit. Dans les 90 minutes suivant l’injection, tous les volontaires ont développé une réponse inflammatoire systémique caractérisée par une libération massive de cytokines pro-inflammatoires et un tableau clinique incluant céphalées, myalgies, nausées, érythème, diarrhée, vasodilatation et hypotension [15]. Ce tableau clinique s’est ensuite aggravé dans les 12 à 24 heures avec la présence d’infiltrats pulmonaires, d’une insuffisance rénale aiguë et d’une coagulation intravasculaire disséminée. Dans les 8 heures après l’injection, tous les volontaires ont développé une lymphopénie sévère et dès la première heure une augmentation très importante des taux sériques de TNF-α, suivie d’une élévation des concentrations circulantes d’IFN-γ et d’IL(interleukine)-10 a été observée. Plusieurs équipes ont récemment essayé d’élucider le mécanisme de ce choc cytokinique présent chez l’homme mais absent dans des conditions similaires chez le singe cynomolgus recevant le TGN1412. Stebbings et al. ont testé 6 protocoles différents de stimulation des lymphocytes périphériques humains ou de singes in vitro. Deux conditions seulement induisent la production de TNF-α, d’IL-6 et d’IL-8 par les cellules humaines mais pas par les cellules de singe : l’adsorption du TGN1412 sur les puits de culture après séchage et l’addition sous forme soluble du TGN1412 en présence à la fois de lymphocytes et de cellules endothéliales humaines [16]. Müller et al. ont montré en employant le JJ316 (équivalent du TGN1412 chez le rat), la présence de deux vagues d’activation des lymphocytes T [17]. La première vague est caractérisée par la sécrétion d’IL-17, d’IFN-γ et de TNF-α et une redistribution rapide des lymphocytes CD4+ de la périphérie vers la rate et les ganglions lymphatiques. La deuxième vague correspond à l’expansion d’une population de lymphocytes T régulateurs CD4+CD25+FoxP3+. Néanmoins, les signes cliniques décrits chez l’homme sont absents chez le rat et la quantité de cytokines circulantes est aussi bien inférieure chez le rat que chez l’homme. Une des hypothèses avancées pour expliquer cette différence inter-espèces serait une sécrétion importante de cytokines chez l’homme par rapport au rat et non pas un effet sur la synthèse même de ces cytokines.
Toxicité non liée à la cible
Immunogénicité
L’immunogénicité des anticorps thérapeutiques peut constituer un problème important en clinique avec l’apparition d’effets toxiques souvent liés à des phénomènes d’hypersensibilité (→). L’immunogénicité d’un anticorps dépend de son contenu en séquences non humaines. Les anticorps murins sont les plus immunogéniques, ce qui limite généralement leur emploi à des traitements de courte durée. L’arrivée sur le marché des anticorps chimériques, humanisés, et maintenant complètement humains, a considérablement réduit l’immunogénicité des anticorps thérapeutiques (→). Parmi les effets toxiques décrits, on trouve des chocs anaphylactiques liés à la production d’IgE anti-basiliximab (anti-CD25) ou après traitement par l’infliximab [18].
(→) voir P. Stas et I. Lasters, page 1070
(→) voir M. Cogné et al., page 1149
Un autre problème lié à l’immunogénicité des anticorps thérapeutiques est une augmentation de leur vitesse d’élimination du fait de la formation de complexes immuns rapidement éliminés par le système réticulo-endothélial. Chez la souris, l’injection d’un anticorps anti-idiotype spécifique d’un anticorps administré précédemment augmente la clairance de celui-ci de manière dose-dépendante (→). Ce résultat suggère que malgré l’absence de séquences non humaines, il y aura toujours une réponse immunogénique liée à l’injection d’anticorps thérapeutiques [19] du fait de la présence d’épitopes immunogènes sur les régions hypervariables.
(→) voir G. Paintaud, page 1057
Détection des effets dans les études précliniques
Les spécialités pharmaceutiques doivent être évaluées pour leur toxicité avant leur mise sur le marché. Cette évaluation consiste en des études conduites majoritairement chez l’animal et visant à identifier le danger lié à la molécule : identification des organes cibles, risque mutagène et cancérigène, toxicité sur les fonctions de reproduction, immunotoxicité et effets sur les grandes fonctions vitales. Certaines de ces études sont effectuées avant l’entrée dans les essais cliniques.
Les effets toxiques décrits ci-dessus, et surtout l’accident récent du TGN1412, montrent bien que la relative confiance en termes de sécurité d’emploi liée à l’utilisation des anticorps en thérapeutique n’est plus d’actualité. L’EMEA (European medicines agency) a d’ailleurs publié le 1er septembre 2007 une nouvelle ligne directrice (Guideline on strategies to identify and mitigate risks for first in human clinical trials with investigational medicinal products) visant à faciliter la transition des modèles non cliniques vers les premiers essais cliniques chez l’homme [20] (→). Une meilleure connaissance des cibles de ces anticorps et de leurs mécanismes d’action et l’identification des facteurs de risque (activation des cellules système immunitaire, cibles pléïotropes…) doit permettre d’améliorer la sécurité d’emploi de ces produits en clinique.
(→) voir F. Lackner et M.E. Behr-Gross, page 1183
Conflit D’Intérêts
L’auteur déclare n’avoir aucun conflit d’intérêts concernant les données publiées dans cet article.
Références
- Gerber HP, Ferrara N. Pharmacology and pharmacodynamics of bevacizumab as monotherapy or in combination with cytotoxic therapies in preclinical studies. Cancer Res 2005; 65 : 671–80. [Google Scholar]
- Hurwitz H, Saini S. Bevacizumab in the treatment of metastatic colorectal cancer: safety profile and management of adverse events. Semin Oncol 2006; 33 : S26–34. [Google Scholar]
- Hodi FS, Butler M, Oble DA, et al. Immunologic and clinical effects of antibody blockade of cytotoxic T lymphocyte-associated antigen 4 in previously vaccinated cancer patients. Proc Natl Acad Sci USA 2008; 105 : 3005–10. [Google Scholar]
- Beck KE, Blansfield JA, Tran KQ, et al. Enterocolitis in patients with cancer after antibody blockade of cytotoxic T-lymphocyte-associated antigen 4. J Clin Oncol 2006; 24 : 2283–9. [Google Scholar]
- Tabrizi MA, Roskos LK. Preclinical and clinical safety of monoclonal antibodies. Drug Discov Today 2007; 12 : 540–7. [Google Scholar]
- Saif MW, Kim R. Incidence and management of cutaneous toxicities associated with cetuximab. Expert Opin Drug Saf 2007; 6 : 175–82. [Google Scholar]
- Cook-Bruns, R. Retrospective analysis of the safety of Herceptin immunotherapy in metastatic breast cancer. Oncology 2001; 61 : 58–66. [Google Scholar]
- Bria E, Cuppone F, Milella M, et al. Trastuzumab cardiotoxicity: biological hypotheses and clinical open issues. Expert Opin Biol Ther 2008; 8 : 1963–71. [Google Scholar]
- de Korte MA, de Vries EG, Lub-de Hooge MN, et al. 111Indium-trastuzumab visualises myocardial human epidermal growth factor receptor 2 expression shortly after anthracycline treatment but not during heart failure: a clue to uncover the mechanisms of trastuzumab-related cardiotoxicity. Eur J Cancer 2007; 43 : 2046–51. [Google Scholar]
- Winkler U, Jensen M, Manzke O, et al. Cytokine-release syndrome in patients with B-cell chronic lymphocytic leukemia and high lymphocyte counts after treatment with an anti-CD20 monoclonal antibody (rituximab, IDEC-C2B8). Blood 1999; 94 : 2217–24. [Google Scholar]
- Wing MG, Moreau T, Greenwood J, et al. Mechanism of first-dose cytokine-release syndrome by CAMPATH 1-H: involvement of CD16 (FcgammaRIII) and CD11a/CD18 (LFA-1) on NK cells. J Clin Invest 1996; 98 : 2819–26. [Google Scholar]
- Chatenoud L, Ferran C, Legendre C, et al. In vivo cell activation following OKT3 administration. Systemic cytokine release and modulation by corticosteroids. Transplantation 1990; 49 : 697–702. [Google Scholar]
- Suntharalingam G, Perry MR, Ward S, et al. Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N Engl J Med 2006; 355 : 1018–28. [Google Scholar]
- Chatenoud L, Bluestone JA. CD3-specific antibodies: a portal to the treatment of autoimmunity. Nat Rev Immunol 2007; 7 : 622–32. [Google Scholar]
- Suntharalingam G, Perry MR, Ward S, et al. Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N Engl J Med 2006; 355 : 1018–28. [Google Scholar]
- Stebbings R, Findlay L, Edwards C, et al. Cytokine storm in the phase I trial of monoclonal antibody TGN1412: better understanding the causes to improve preclinical testing of immunotherapeutics. J Immunol 2007; 179 : 3325–31. [Google Scholar]
- Müller N, van den Brandt J, Odoardi F, et al. A CD28 superagonistic antibody elicits 2 functionally distinct waves of T cell activation in rats. J Clin Invest 2008; 118 : 1405–16. [Google Scholar]
- Baudouin V, Crusiaux A, Haddad E, et al. Anaphylactic shock caused by immunoglobulin E sensitization after retreatment with the chimeric anti-interleukin-2 receptor monoclonal antibody basiliximab. Transplantation 2003; 76 : 459–63. [Google Scholar]
- Johansson A, Erlandsson A, Eriksson D, et al. Idiotypic-anti-idiotypic complexes and their in vivo metabolism. Cancer 2002; 94 : 1306–13. [Google Scholar]
- Committee for medicinal products for human use. European Medecines Agency. Guideline on strategies to identify and mitigate risks for first in human clinical trials with investigational medicinal products. September 1st 2007. http://www.emea.europa.eu/pdfs/human/swp/2836707enfin.pdf [Google Scholar]
Aucun commentaire:
Enregistrer un commentaire
Remarque : Seul un membre de ce blog est autorisé à enregistrer un commentaire.