Les documents hautement confidentiels de Pfizer, qui ont été synonymes de l'extrême manque de transparence révélé par les actions des principales agences gouvernementales, au cours des 20 derniers mois, amenant les critiques du récit officiel à demander "montrez-nous les données", sont enfin révélés - enfin en quelque sorte : les quelques premières centaines de pages expurgées sur un total de 451 000.
Qu'est-ce qui a conduit à cette divulgation ?
La faille dans le dispositif de sécurité draconien de Pfizer et de la Food and Drug Administration (FDA) est apparue sous la forme d'un communiqué de la Freedom of Information Act (FOIA) avec la demande déposée le 27 août 2021 pour accéder à toute la documentation de Pfizer sur laquelle la FDA s'était appuyée pour autoriser l'utilisation en urgence du vaccin Pfizer-BioNTech Covid-19. Une agence qui a reçu une requête de FOIA est tenue de "déterminer, dans les 20 jours ouvrables suivant la réception de cette demande, s'il convient d'y donner suite", comme le prévoit la loi de 1967 sur le droit à l'information. Il a pourtant fallu trois mois à la FDA pour publier les 91 premières pages caviardées, le 20 novembre.
La requête de FOIA a été formulée par un groupe de plus de 30 scientifiques et universitaires qui ont intenté une action en justice contre l'agence parce qu'elle n'avait pas entièrement satisfait à la demande, puisque moins de 1 % de la documentation avait été divulgué et que la FDA avait déclaré que toutes les données seraient partagées d'ici 2076. Par la suite, l'agence gouvernementale a eu l'audace de repousser la date encore plus loin, à 2096. Cela en raison de sa révélation de l'existence de milliers de pages supplémentaires, soit 451 000 pages au total, contre 320 000 pages initialement prévues. Toutefois, le rythme auquel la FDA est disposée à publier la documentation n'a pas changé et reste de 500 pages par mois. Il convient de noter qu'il n'a fallu que 108 jours à la FDA pour examiner toute la documentation de Pfizer avant d'autoriser l'utilisation du vaccin Pfizer BNT162B2 dans le cadre d'une autorisation d'urgence le 1er décembre 2020.
La faille dans le dispositif de sécurité draconien de Pfizer et de la Food and Drug Administration (FDA) est apparue sous la forme d'un communiqué de la Freedom of Information Act (FOIA) avec la demande déposée le 27 août 2021 pour accéder à toute la documentation de Pfizer sur laquelle la FDA s'était appuyée pour autoriser l'utilisation en urgence du vaccin Pfizer-BioNTech Covid-19. Une agence qui a reçu une requête de FOIA est tenue de "déterminer, dans les 20 jours ouvrables suivant la réception de cette demande, s'il convient d'y donner suite", comme le prévoit la loi de 1967 sur le droit à l'information. Il a pourtant fallu trois mois à la FDA pour publier les 91 premières pages caviardées, le 20 novembre.
La requête de FOIA a été formulée par un groupe de plus de 30 scientifiques et universitaires qui ont intenté une action en justice contre l'agence parce qu'elle n'avait pas entièrement satisfait à la demande, puisque moins de 1 % de la documentation avait été divulgué et que la FDA avait déclaré que toutes les données seraient partagées d'ici 2076. Par la suite, l'agence gouvernementale a eu l'audace de repousser la date encore plus loin, à 2096. Cela en raison de sa révélation de l'existence de milliers de pages supplémentaires, soit 451 000 pages au total, contre 320 000 pages initialement prévues. Toutefois, le rythme auquel la FDA est disposée à publier la documentation n'a pas changé et reste de 500 pages par mois. Il convient de noter qu'il n'a fallu que 108 jours à la FDA pour examiner toute la documentation de Pfizer avant d'autoriser l'utilisation du vaccin Pfizer BNT162B2 dans le cadre d'une autorisation d'urgence le 1er décembre 2020.
Les scientifiques, les responsables de la santé publique et les universitaires, dirigés par le Dr Peter McCullough, forment le groupe de plaignants, PHMPT (Public Health and Medical Professionals for Transparency) et sont représentés par le cabinet d'avocats Aaron Siri, de Siri & Glimstad LLP.
Dans une interview exclusive accordée à Trial Site News, Aaron Siri, associé directeur du cabinet, qui a une grande expérience des litiges civils, a déclaré :
"Le tribunal n'a pas encore ordonné la production d'une seule page. Dans la plupart des cas, lorsque notre cabinet soumet une demande en vertu de la loi sur le droit à l'information, ils [l'agence] produisent des documents, mais la FDA veut le faire à un rythme incroyablement lent, qui ne correspond pas aux besoins de la demande. Le problème n'est pas de savoir s'ils vont produire les documents - ils vont les produire. Le problème est de savoir combien de temps cela prendra et, une fois que nous l'aurons obtenu, le problème sera celui des modifications qu'ils y apporteront."Lorsque je lui ai demandé si des requêtes de FOIA seront faites pour obtenir les documents de Moderna et de Janssen (une filiale de Johnson and Johnson) fournis à la FDA pour obtenir l'autorisation d'utilisation en urgence, il a répondu :
"Vous ne pouvez pas faire de demande tant qu'un vaccin n'a pas été homologué. L'autorisation d'utilisation d'urgence n'est pas la même chose que l'homologation ou l'approbation. Le vaccin Pfizer est le seul vaccin qui a été homologué/approuvé comme étant "sûr et efficace" selon la FDA le 23 août 2021."Vous trouverez des détails sur l'affaire et les documents judiciaires pertinents sur le blog d'Aaron Siri, Injecting Freedom.
Les premières centaines de pages des documents Pfizer récemment publiés ont été partagées sur le site Web du PHMPT.
Le présent rapport d'enquête se concentre sur le document de 38 pages intitulé "Cumulative Analysis of Post-Authorization Adverse Event Reports of PF-07302048 (BNT162B2) received through 28 February 2021 ". Le rapport a été préparé par Pfizer, entre le 1er décembre 2020 et le 28 février 2021. Les rapports d'événements indésirables proviennent des États-Unis, du Royaume-Uni, d'Italie, d'Allemagne, de France, du Portugal, d'Espagne et de 56 autres pays.
Il est intéressant de noter que l'artefact représente une analyse modifiée fournie par Pfizer, en réponse à ses manquements associés à la soumission incomplète d'un ensemble de données de sécurité à la FDA, sur laquelle l'agence a émis des commentaires. Une référence est faite à la demande du 9 mars de la FDA à Pfizer : "Nous sommes très intéressés par une analyse cumulative des données de sécurité post-autorisation pour soutenir votre future soumission BLA. Veuillez soumettre une analyse intégrée de vos données cumulatives de sécurité post-autorisation, y compris l'expérience post-autorisation aux États-Unis et à l'étranger, dans votre prochaine soumission de BLA. Veuillez inclure une analyse cumulative des risques importants identifiés, des risques potentiels importants et des zones d'informations manquantes importantes identifiés dans votre plan de pharmacovigilance, ainsi que des événements indésirables d'intérêt particulier et des erreurs d'administration du vaccin (associées ou non à un événement indésirable). Veuillez également inclure les données de distribution et une analyse des événements indésirables les plus courants. En outre, veuillez soumettre votre plan de pharmacovigilance mis à jour avec votre demande de BLA.
Les nombreuses inconnues
Au cours de la courte période de trois mois pendant laquelle les données ont été "signalées spontanément à Pfizer", 42 086 cas ont été enregistrés avec 158 893 événements. D'après les données, on peut interpréter que la personne moyenne (cas) aurait souffert d'un peu moins de quatre symptômes (événements). Fait particulièrement troublant, la FDA a choisi de protéger les intérêts de Pfizer en caviardant le nombre total de doses à (b) (4), empêchant ainsi de calculer les taux d'incidence et de fournir une analyse significative des données. Un autre fait profondément inquiétant concerne des limitations importantes citées par Pfizer : "l'ampleur de la sous-déclaration est inconnue". À ce sujet, les chercheurs qui ont dirigé une importante étude de Harvard menée de 2007 à 2010 ont découvert que "moins de 0,3 % de tous les effets indésirables des médicaments et de 1 à 13 % des effets graves sont signalés à la FDA". En supposant que ce calcul soit correct, nous pouvons conclure que les 42 086 cas représentent un nombre stupéfiant de cas non signalés.
D'autres " inconnues " significatives parsèment l'analyse de Pfizer :
- 2.990 cas où le sexe est inconnu
- 6.876 cas où l'âge est inconnu
- 9.440 cas dont l'issue est inconnue
Une autre anomalie est que, pour les issues concernant les cas, Pfizer a choisi d'inclure les personnes qui se rétablissent d'événements indésirables dans la même catégorie que celles qui sont rétablies, sous l'étiquette "rétabli/en cours de rétablissement". Cette décision à elle seule semble discutable.
Le grand nombre de déclarations spontanées d'événements indésirables
Il est alarmant de constater que l'analyse fait état d'un tel volume d'événements indésirables, classés comme "cas graves", au cours de cette courte période, que Pfizer a dû embaucher davantage d'employés à temps plein et procéder à des changements technologiques importants pour pouvoir traiter les rapports volumineux tout en respectant les délais réglementaires. Comme indiqué dans le document :
Les 1.228 décès
Dans le document généré par Pfizer, un sérieux signal d'alarme apparaît : 1.228 personnes sont décédées dans les trois mois suivant la prise du vaccin, alors qu'aucun enregistrement ne rend compte du sexe des participants à l'étude qui sont décédés. Ces données, qui ont d'importantes répercussions sur la sécurité, étaient connues de Pfizer à la fin du mois de février. Pourtant, le 12 avril, le Dr Mace Rothenberg, ancien directeur médical de Pfizer, a déclaré au Washington Journal à propos de la mise au point du vaccin Pfizer : "Je peux vous dire qu'aucun détail n'a été négligé" et "aucun décès n'a été directement imputé au seul vaccin". Ceux qui défendent la sécurité du vaccin Pfizer ont soulevé l'argument selon lequel " la corrélation n'implique pas la causalité, c'est-à-dire que deux événements qui se produisent ensemble n'établissent pas une relation de cause à effet ".
Le grand nombre de déclarations spontanées d'événements indésirables
Il est alarmant de constater que l'analyse fait état d'un tel volume d'événements indésirables, classés comme "cas graves", au cours de cette courte période, que Pfizer a dû embaucher davantage d'employés à temps plein et procéder à des changements technologiques importants pour pouvoir traiter les rapports volumineux tout en respectant les délais réglementaires. Comme indiqué dans le document :
En raison du grand nombre de déclarations spontanées d'effets indésirables reçues pour le produit, le titulaire de l'autorisation de mise sur le marché a donné la priorité au traitement des cas graves, afin de respecter les délais de déclaration réglementaire et de s'assurer que ces déclarations sont disponibles pour la détection des signaux et les activités d'évaluation. Le rapport indique ensuite comment Pfizer a traité ce grand nombre de rapports d'événements indésirables. Pfizer a également pris de nombreuses mesures pour atténuer la forte augmentation du nombre de rapports d'événements indésirables. Ces mesures comprennent des améliorations technologiques importantes, des solutions en matière de processus et de flux de travail, ainsi que l'augmentation du nombre de collègues chargés de la saisie des données et du traitement des cas. À ce jour, Pfizer a embauché b4 employés à temps plein (ETP) supplémentaires...(*b4 est un terme expurgé)
Les 1.228 décès
Dans le document généré par Pfizer, un sérieux signal d'alarme apparaît : 1.228 personnes sont décédées dans les trois mois suivant la prise du vaccin, alors qu'aucun enregistrement ne rend compte du sexe des participants à l'étude qui sont décédés. Ces données, qui ont d'importantes répercussions sur la sécurité, étaient connues de Pfizer à la fin du mois de février. Pourtant, le 12 avril, le Dr Mace Rothenberg, ancien directeur médical de Pfizer, a déclaré au Washington Journal à propos de la mise au point du vaccin Pfizer : "Je peux vous dire qu'aucun détail n'a été négligé" et "aucun décès n'a été directement imputé au seul vaccin". Ceux qui défendent la sécurité du vaccin Pfizer ont soulevé l'argument selon lequel " la corrélation n'implique pas la causalité, c'est-à-dire que deux événements qui se produisent ensemble n'établissent pas une relation de cause à effet ".
La page 10 de l'analyse de Pfizer présente un important risque identifié d'anaphylaxie avec neuf décès signalés. Quatre de ces neuf décès sont survenus le jour même où les personnes ont été vaccinées (voir ci-dessous).
Pfizer a souligné que ces personnes souffraient d'affections sous-jacentes, mais le fait qu'elles soient toutes les quatre décédées le jour même où elles ont reçu le vaccin suggère une causalité potentielle de décès vaccinal.
Dans le tableau 7 des pages 16 et 17, 1403 cas d'EIAS (événement indésirable d'intérêt spécifique) cardiovasculaires ont été signalés et segmentés comme suit : Arythmie ; Insuffisance cardiaque ; Insuffisance cardiaque aiguë ; Choc cardiogénique ; Coronaropathie ; Infarctus du myocarde ; Syndrome de tachycardie orthostatique posturale ; Cardiomyopathie d'effort ; Tachycardie.
La latence d'apparition de l'événement pertinent allait de moins de 24 heures à 21 jours. Cela signifie que les événements pertinents sont survenus à n'importe quel moment entre moins de 24 heures et 21 jours après la réception du vaccin, avec une médiane de moins de 24 heures. 136 événements pertinents ont eu une issue fatale. Par conséquent, 50 % de ces événements pertinents (y compris les décès) sont survenus moins de 24 heures après l'administration du vaccin. Cela indique une fois de plus la causalité de décès vaccinal.
Pourtant, Pfizer conclut : "Cette revue des cas cumulés ne soulève pas de nouveaux problèmes de sécurité. La surveillance va se poursuivre".
Si l'on examine la catégorie "IESA à médiation immunitaire/auto-immune", 1 050 cas ont été signalés, avec un peu plus de trois fois plus de femmes affectées que d'hommes - il y a eu 12 issues fatales. La médiane de la latence d'apparition de l'événement pertinent était inférieure à 24 heures, ce qui suggère à nouveau une causalité de décès vaccinal.
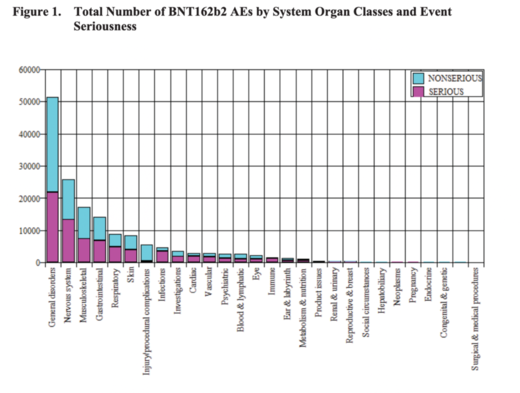
La gravité des cas
Si l'on regarde le graphique ci-dessous, on constate qu'une part importante des cas sont déclarés comme étant graves par rapport aux cas non graves, le nombre le plus élevé de cas graves se trouvant dans la catégorie "troubles généraux". Un cas grave est un cas qui est médicalement important et qui entraîne soit une hospitalisation, soit une conséquence mettant la vie en danger, soit le décès. Il est intéressant de noter que pour les troubles cardiaques, immunitaires, vasculaires et les infections, les cas graves dominent et que pour les troubles immunitaires, tous sont classés comme graves.
Dans le tableau 7 des pages 16 et 17, 1403 cas d'EIAS (événement indésirable d'intérêt spécifique) cardiovasculaires ont été signalés et segmentés comme suit : Arythmie ; Insuffisance cardiaque ; Insuffisance cardiaque aiguë ; Choc cardiogénique ; Coronaropathie ; Infarctus du myocarde ; Syndrome de tachycardie orthostatique posturale ; Cardiomyopathie d'effort ; Tachycardie.
La latence d'apparition de l'événement pertinent allait de moins de 24 heures à 21 jours. Cela signifie que les événements pertinents sont survenus à n'importe quel moment entre moins de 24 heures et 21 jours après la réception du vaccin, avec une médiane de moins de 24 heures. 136 événements pertinents ont eu une issue fatale. Par conséquent, 50 % de ces événements pertinents (y compris les décès) sont survenus moins de 24 heures après l'administration du vaccin. Cela indique une fois de plus la causalité de décès vaccinal.
Pourtant, Pfizer conclut : "Cette revue des cas cumulés ne soulève pas de nouveaux problèmes de sécurité. La surveillance va se poursuivre".
Si l'on examine la catégorie "IESA à médiation immunitaire/auto-immune", 1 050 cas ont été signalés, avec un peu plus de trois fois plus de femmes affectées que d'hommes - il y a eu 12 issues fatales. La médiane de la latence d'apparition de l'événement pertinent était inférieure à 24 heures, ce qui suggère à nouveau une causalité de décès vaccinal.
La gravité des cas
Si l'on regarde le graphique ci-dessous, on constate qu'une part importante des cas sont déclarés comme étant graves par rapport aux cas non graves, le nombre le plus élevé de cas graves se trouvant dans la catégorie "troubles généraux". Un cas grave est un cas qui est médicalement important et qui entraîne soit une hospitalisation, soit une conséquence mettant la vie en danger, soit le décès. Il est intéressant de noter que pour les troubles cardiaques, immunitaires, vasculaires et les infections, les cas graves dominent et que pour les troubles immunitaires, tous sont classés comme graves.
Les femmes étaient x3 fois plus touchées par les effets indésirables du vaccin
Dans toutes les catégories d'événements indésirables d'intérêt particulier, les femmes sont généralement trois fois plus touchées que les hommes. Toutefois, ce phénomène n'était nulle part aussi prononcé que dans le cas de l'anaphylaxie (une réaction allergique potentiellement mortelle), où les femmes étaient plus de huit fois plus touchées. Sur les 1002 cas d'anaphylaxie rapportés répondant au niveau 1-4 de la Brighton Collaboration (le niveau 1 étant le plus haut niveau de certitude diagnostique de l'anaphylaxie), 876 femmes ont été affectées contre 106 hommes. Les femmes étaient également beaucoup plus touchées par les événements cardiovasculaires ; 1076 femmes ont été signalées comme des cas contre 291 hommes. Ces données statistiquement significatives révèlent la possibilité réelle de risques liés à la sécurité des vaccins en fonction du sexe.
Pourtant, Pfizer ne commente nulle part ces données dans son analyse, mais réaffirme avec assurance que "l'examen des cas cumulés ne soulève aucun nouveau problème de sécurité".
Les informations manquantes
Il convient également de noter que les données associées à la rubrique "Utilisation pendant la grossesse et l'allaitement" ont été on ne sait pourquoi exclues de l'analyse initiale soumise à la FDA. Dans la version modifiée, 413 cas d'effets indésirables sont signalés, dont 84 sont classés comme graves.
Les résultats des 270 grossesses ont été rapportés comme suit : avortement spontané (23), résultat en attente (5), naissance prématurée avec décès néonatal, avortement spontané avec décès intra-utérin (2 chacun), avortement spontané avec décès néonatal et résultat normal (1 chacun).
Il est alarmant que Pfizer affirme que "l'examen de ces cas d'utilisation pendant la grossesse et l'allaitement n'a fait apparaître aucun signal de sécurité". Les données contenues dans le document fortement expurgé semblent contredire cette évaluation optimiste.
Chez les enfants de moins de 12 ans, qui à l'origine ne figuraient pas dans l'analyse de Pfizer, 34 cas ont été signalés, dont 24 ont été classés comme graves. Le fait que de jeunes enfants aient reçu le vaccin de Pfizer suscite des inquiétudes, puisque l'entreprise n'a pas reçu d'autorisation d'utilisation d'urgence pour administrer le vaccin à la population pédiatrique à ce moment-là. En outre, la fourchette d'âge a suscité une inquiétude considérable, car elle allait de 2 mois à 9 ans. Le rapport ne contient pas de données sur le nombre total d'enfants ayant reçu le vaccin. Il n'est donc pas possible de calculer les taux d'incidence pour extrapoler une analyse significative.
Un exemple de liste des IESA connus dans l'analyse cumulative de Pfizer
Affections hématologiques et du système lymphatique : Lymphadénopathie
Événements cardiovasculaires : infarctus aigu du myocarde ; Arythmie ; Insuffisance cardiaque ; Insuffisance cardiaque aiguë ; Choc cardiogénique; Maladie de l'artère coronaire; Infarctus du myocarde; Syndrome de tachycardie orthostatique posturale ; Cardiomyopathie d'effort ; Tachycardie
Problèmes gastro-intestinaux
Troubles généraux et anomalies au site d'administration
Infections et infestations
Troubles musculo-squelettiques et du tissu conjonctif : Arthralgie; Arthrite; Arthrite bactérienne; Syndrome de fatigue chronique; polyarthrite; Polyneuropathie; Syndrome de fatigue post-virale ; La polyarthrite rhumatoïde
Troubles du système nerveux
Troubles respiratoires, thoraciques et médiastinaux : infections des voies respiratoires inférieures ; insuffisances respiratoires, infections virales des voies respiratoires inférieures; syndrome de détresse respiratoire aiguë; Intubation endotrachéale; Hypoxie ; Hémorragie pulmonaire ; Trouble respiratoire ; Syndrome respiratoire aigu sévère
Affections de la peau et du tissu sous-cutané
Anaphylaxie
Maladie augmentée associée au vaccin (VAED) comprenant la maladie respiratoire augmentée associée au vaccin (VAERD).
COVID-19
Paralysie faciale
Troubles à médiation immunitaire/auto-immuns
Neurologique (y compris la démyélinisation) : Convulsions ; Ataxie; Cataplexie ; Encéphalopathie ; Fibromyalgie ; Augmentation de la pression intracrânienne ; Méningite; Méningite aseptique ; Narcolepsie
Liés à la grossesse : infection de la cavité amniotique ; Césarienne; Anomalie congénitale; Décès néonatal; Éclampsie; syndrome de détresse fœtale ; Bébé de faible poids à la naissance ; Exposition maternelle pendant la grossesse ; placenta praevia; Pré-éclampsie ; Accouchement prématuré; Mortinaissance ; Rupture utérine ; Vasa praevia
Rénal : Lésion rénale aiguë, insuffisance rénale
Événements thromboemboliques : embolie et thrombose ; AVC AESI, thrombose veineuse profonde ; Coagulation intravasculaire disséminée; Embolie; Embolie veineuse ; Embolie pulmonaire
Cela vaut la peine de comparer la liste ci-dessus avec la liste ci-dessous accessible via le site Web de la FDA sous le document « fiche d'information Pfizer-BioNTech pour les destinataires et les soignants », révisée au 9 décembre 2021. Il est manifeste que de nombreux effets secondaires engageant le pronostic vital n'ont pas été inclus, même si l'analyse cumulative de Pfizer des rapports d'événements indésirables post-autorisation a été produite pour la FDA le 30 avril 2021.
Dans toutes les catégories d'événements indésirables d'intérêt particulier, les femmes sont généralement trois fois plus touchées que les hommes. Toutefois, ce phénomène n'était nulle part aussi prononcé que dans le cas de l'anaphylaxie (une réaction allergique potentiellement mortelle), où les femmes étaient plus de huit fois plus touchées. Sur les 1002 cas d'anaphylaxie rapportés répondant au niveau 1-4 de la Brighton Collaboration (le niveau 1 étant le plus haut niveau de certitude diagnostique de l'anaphylaxie), 876 femmes ont été affectées contre 106 hommes. Les femmes étaient également beaucoup plus touchées par les événements cardiovasculaires ; 1076 femmes ont été signalées comme des cas contre 291 hommes. Ces données statistiquement significatives révèlent la possibilité réelle de risques liés à la sécurité des vaccins en fonction du sexe.
Pourtant, Pfizer ne commente nulle part ces données dans son analyse, mais réaffirme avec assurance que "l'examen des cas cumulés ne soulève aucun nouveau problème de sécurité".
Les informations manquantes
Il convient également de noter que les données associées à la rubrique "Utilisation pendant la grossesse et l'allaitement" ont été on ne sait pourquoi exclues de l'analyse initiale soumise à la FDA. Dans la version modifiée, 413 cas d'effets indésirables sont signalés, dont 84 sont classés comme graves.
Les résultats des 270 grossesses ont été rapportés comme suit : avortement spontané (23), résultat en attente (5), naissance prématurée avec décès néonatal, avortement spontané avec décès intra-utérin (2 chacun), avortement spontané avec décès néonatal et résultat normal (1 chacun).
Il est alarmant que Pfizer affirme que "l'examen de ces cas d'utilisation pendant la grossesse et l'allaitement n'a fait apparaître aucun signal de sécurité". Les données contenues dans le document fortement expurgé semblent contredire cette évaluation optimiste.
Chez les enfants de moins de 12 ans, qui à l'origine ne figuraient pas dans l'analyse de Pfizer, 34 cas ont été signalés, dont 24 ont été classés comme graves. Le fait que de jeunes enfants aient reçu le vaccin de Pfizer suscite des inquiétudes, puisque l'entreprise n'a pas reçu d'autorisation d'utilisation d'urgence pour administrer le vaccin à la population pédiatrique à ce moment-là. En outre, la fourchette d'âge a suscité une inquiétude considérable, car elle allait de 2 mois à 9 ans. Le rapport ne contient pas de données sur le nombre total d'enfants ayant reçu le vaccin. Il n'est donc pas possible de calculer les taux d'incidence pour extrapoler une analyse significative.
Un exemple de liste des IESA connus dans l'analyse cumulative de Pfizer
Affections hématologiques et du système lymphatique : Lymphadénopathie
Événements cardiovasculaires : infarctus aigu du myocarde ; Arythmie ; Insuffisance cardiaque ; Insuffisance cardiaque aiguë ; Choc cardiogénique; Maladie de l'artère coronaire; Infarctus du myocarde; Syndrome de tachycardie orthostatique posturale ; Cardiomyopathie d'effort ; Tachycardie
Problèmes gastro-intestinaux
Troubles généraux et anomalies au site d'administration
Infections et infestations
Troubles musculo-squelettiques et du tissu conjonctif : Arthralgie; Arthrite; Arthrite bactérienne; Syndrome de fatigue chronique; polyarthrite; Polyneuropathie; Syndrome de fatigue post-virale ; La polyarthrite rhumatoïde
Troubles du système nerveux
Troubles respiratoires, thoraciques et médiastinaux : infections des voies respiratoires inférieures ; insuffisances respiratoires, infections virales des voies respiratoires inférieures; syndrome de détresse respiratoire aiguë; Intubation endotrachéale; Hypoxie ; Hémorragie pulmonaire ; Trouble respiratoire ; Syndrome respiratoire aigu sévère
Affections de la peau et du tissu sous-cutané
Anaphylaxie
Maladie augmentée associée au vaccin (VAED) comprenant la maladie respiratoire augmentée associée au vaccin (VAERD).
COVID-19
Paralysie faciale
Troubles à médiation immunitaire/auto-immuns
Neurologique (y compris la démyélinisation) : Convulsions ; Ataxie; Cataplexie ; Encéphalopathie ; Fibromyalgie ; Augmentation de la pression intracrânienne ; Méningite; Méningite aseptique ; Narcolepsie
Liés à la grossesse : infection de la cavité amniotique ; Césarienne; Anomalie congénitale; Décès néonatal; Éclampsie; syndrome de détresse fœtale ; Bébé de faible poids à la naissance ; Exposition maternelle pendant la grossesse ; placenta praevia; Pré-éclampsie ; Accouchement prématuré; Mortinaissance ; Rupture utérine ; Vasa praevia
Rénal : Lésion rénale aiguë, insuffisance rénale
Événements thromboemboliques : embolie et thrombose ; AVC AESI, thrombose veineuse profonde ; Coagulation intravasculaire disséminée; Embolie; Embolie veineuse ; Embolie pulmonaire
Cela vaut la peine de comparer la liste ci-dessus avec la liste ci-dessous accessible via le site Web de la FDA sous le document « fiche d'information Pfizer-BioNTech pour les destinataires et les soignants », révisée au 9 décembre 2021. Il est manifeste que de nombreux effets secondaires engageant le pronostic vital n'ont pas été inclus, même si l'analyse cumulative de Pfizer des rapports d'événements indésirables post-autorisation a été produite pour la FDA le 30 avril 2021.

Conclusion
Bien que l'auteur s'efforce de rester aussi objectif et impartial que possible, un examen approfondi de ce seul rapport suggère que la FDA et Pfizer semblent avoir dissimulé au public toute l'étendue des effets secondaires du vaccin Pfizer-BioNTech. Si cette hypothèse est effectivement vraie, alors l'agence de réglementation "Gold Standard" et la prestigieuse multinationale pharmaceutique ont jeté par la fenêtre tout le concept de consentement éclairé.
Cela relève également du simulacre que des mois plus tard, la FDA ait traîné les pieds et ait publié cet important document de sécurité basé sur les rapports de cas d'événements indésirables en vertu de la loi FOIA. Les rapports de cas jouent un rôle important dans la pharmacovigilance. La reconnaissance du lien entre la thalidomide administrée aux mères et les malformations de leurs bébés a été déclenchée par un rapport de cas.
Ce qui serait peut-être encore plus dévastateur - et qui tournerait en dérision tout l'intérêt des systèmes de réglementation avancés censés garantir la sécurité du public - serait que si la FDA gagne le litige en cours pour retarder la publication des informations, le public doive attendre encore 75 ans pour avoir accès à toutes les données, ce qui sera alors beaucoup trop tard.
Bien que l'auteur s'efforce de rester aussi objectif et impartial que possible, un examen approfondi de ce seul rapport suggère que la FDA et Pfizer semblent avoir dissimulé au public toute l'étendue des effets secondaires du vaccin Pfizer-BioNTech. Si cette hypothèse est effectivement vraie, alors l'agence de réglementation "Gold Standard" et la prestigieuse multinationale pharmaceutique ont jeté par la fenêtre tout le concept de consentement éclairé.
Cela relève également du simulacre que des mois plus tard, la FDA ait traîné les pieds et ait publié cet important document de sécurité basé sur les rapports de cas d'événements indésirables en vertu de la loi FOIA. Les rapports de cas jouent un rôle important dans la pharmacovigilance. La reconnaissance du lien entre la thalidomide administrée aux mères et les malformations de leurs bébés a été déclenchée par un rapport de cas.
Ce qui serait peut-être encore plus dévastateur - et qui tournerait en dérision tout l'intérêt des systèmes de réglementation avancés censés garantir la sécurité du public - serait que si la FDA gagne le litige en cours pour retarder la publication des informations, le public doive attendre encore 75 ans pour avoir accès à toutes les données, ce qui sera alors beaucoup trop tard.
Traduction : Résistance mondiale. Corrections : Sott.net
Aucun commentaire:
Enregistrer un commentaire
Remarque : Seul un membre de ce blog est autorisé à enregistrer un commentaire.